

北京时间2026年3月13日,黄荷凤/林圣彩院士联合团队在Vita上公布了他们的发现,哺乳动物胎儿在相对低糖的宫内环境中,通过一种独特的蛋白质化学修饰——“乙酰化”,阻断了肝脏细胞对低糖的应激反应通路,从而保障了合成代谢的持续进行与胎儿的正常发育。这一机制不仅是胎儿应对宫内资源限制的生理适应,更可能是人类漫长演化历史中,为应对母体间歇性食物匮乏而锤炼出的深层生存策略。它从分子层面诠释了生命起源的坚韧,并引发了关于生存环境压力如何塑造生命基本程序的哲学思考。

左起:张宸崧、余传金、林圣彩、黄荷凤、丁国莲、张辰颉
01科学发现:胎儿肝脏的“低糖悖论”与乙酰化“开关”
在哺乳动物的胚胎发育过程中,胎儿生长于一个独特的宫内环境。为确保葡萄糖通过胎盘从母体至胎儿单向转运,胎儿血循环中的葡萄糖浓度始终显著低于母体。这是一个经典的生理学梯度。然而,矛盾之处在于,胎儿正处于一生中生长最迅猛的时期之一,其肝脏需要进行旺盛的合成代谢,而合成代谢是高度耗能的,通常需要充沛的能量原料(如葡萄糖)来驱动。
针对这一有趣的生理学悖论,黄荷凤院士团队通过模拟母胎之间的葡萄糖浓度差异,对不同细胞系进行了梯度葡萄糖浓度的培养实验,发现了令人兴奋的实验结果:尽管在生理状态下胎儿的葡萄糖供给相对匮乏,但胎肝来源的细胞系即使在完全无葡萄糖的培养基中,其增殖速率依然维持在较高水平。这一现象提示,胎肝细胞可能具备独特的代谢适应机制。
在绝大多数成年细胞中,低葡萄糖环境会触发一套精密的营养感应与应对机制,主要表现为:
1、AMPK通路激活:葡萄糖缺乏通过FBP-TRPV-v-ATPase轴或者继发能量短缺(AMP水平升高),激活AMPK,AMPK如同细胞的“节能指挥官”,迅速关闭耗能的合成代谢,开启分解代谢以产生应急能量;
2、mTORC1通路抑制:mTORC1是细胞合成代谢的“总开关”,促进蛋白质、脂质合成。低糖状态下,AMPK的激活以及其他非AMPK依赖机制会强烈抑制mTORC1的活性,使细胞进入“节能模式”。
因此,为探究其背后的调控机制,研究团队首先检测了胎肝细胞在葡萄糖缺乏环境下AMPK及mTORC1这两个关键能量感应分子的活性变化。结果发现,尽管葡萄糖匮乏依然能够激活AMPK,但胎肝细胞中mTORC1的活性并未受到抑制,反而维持在较高水平。这种“AMPK激活而mTORC1未被抑制”的独特状态,与成年细胞的经典反应模式截然不同。进一步在人类和小鼠的胎儿肝脏中证实,在低糖条件下,尽管“节能指挥官”AMPK如预期般被激活,但“合成开关”mTORC1却依然保持着活跃状态。这意味着, 胎儿肝脏细胞似乎“忽视”了低糖的营养危机信号,坚持进行合成代谢。这一反常现象,成为了揭开胎肝细胞代谢适应机制的关键切入点,也由此开启了本项研究课题。
是什么帮助胎儿肝脏细胞做出如此勇敢的决定?
借助林圣彩院士团队开发的AMPK溶酶体途径激活药物对胎肝细胞进行干预,结果显示 mTORC1的活性可被显著抑制。这一发现提示,胎肝细胞在低糖环境下之所以能维持mTORC1活性,并非由于其AMPK溶酶体激活通路本身存在缺陷,而是该通路在低糖条件下未被有效触发。由此,两大团队深度合作,将目光聚焦于调控该通路的上游环节——FBP-TRPV-v-ATPase轴的活性状态,深入探索发现,胎肝细胞中一种离子通道蛋白TRPV4是关键所在。
在成年细胞中,低糖会通过一系列信号(例如此时醛缩酶无法与足够多的果糖-1,6-二磷酸(FBP)结合)最终导致TRPV4关闭,进而抑制mTORC1。在胎儿肝脏中,研究团队基于质谱初步锁定多个潜在的修饰位点,随后通过构建位点特异性突变体并结合功能学实验逐一验证,最终确定,TRPV4蛋白的第608位赖氨酸(K608)发生了独特的“乙酰化”修饰。这种翻译后修饰如同一个化学标签,直接阻断了低糖信号对TRPV4的通道抑制作用,使得mTORC1活性得以维持。
这个标签,就像在胎盘哺乳动物漫漫演化长夜中点亮的一盏灯,暂时屏蔽了“低糖的饥饿信号”,让胎儿在母体营养波动时,依然能保持合成代谢的引擎(mTORC1通路)全速运转。
为了验证这一机制的决定性作用,研究团队进行了点突变实验:当在胎儿肝细胞中表达一个无法被乙酰化的TRPV4突变体(K608R)时,细胞恢复了对低糖的正常敏感性,mTORC1被有效抑制。更重要的是,小鼠模型中,在胚胎发育期特异性表达此突变体,会导致胎儿肝脏合成代谢受损、蛋白质合成减少,最终引发胎儿肝脏发育异常甚至胎儿死亡。这充分证明,TRPV4-K608的乙酰化,是胎儿在低糖环境中维持生命建设程序不可中断的核心分子“开关”。
研究团队进一步追问:究竟是谁给胎儿的TRPV4戴上了这道“护身符”,让它能在低糖环境下依然维持合成代谢?通过系统筛选,团队锁定了关键分子——乙酰基转移酶p300。实验发现,如果敲低p300的活性,或者过表达去乙酰化酶SIRT2/SIRT7,都能让胎肝细胞恢复对低糖的敏感性,抑制mTORC1。体外实验进一步证实,p300可以直接给TRPV4的K608位点贴上乙酰基标签。更有意义的是,p300在肝脏中的表达水平在出生后会急剧下降,这与胎儿肝脏的特殊代谢状态在出生后消失的现象完美吻合。这一发现揭示,p300正是那位在胎儿时期为TRPV4戴上“护身符”的“守护者”。
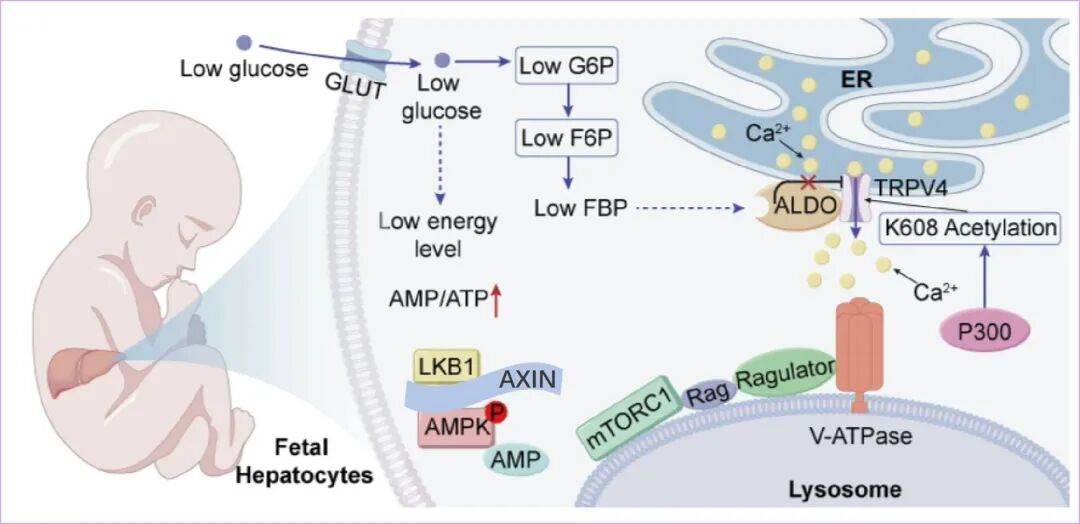
图:胎儿肝脏细胞通过TRPV4乙酰化拮抗低糖诱导的mTORC1抑制
02演化意义:对母体饥饿的古老“预适应”
这一发现远不止于解释一个发育生物学现象,它更指向了一个深刻的演化生物学逻辑:胎儿对宫内低糖环境的适应,可能是对母体在漫长演化史上长期面临食物短缺(饥饿)的一种超前“预适应”。
在人类及其远古祖先的生存史上,怀孕的母亲无法保证持续、稳定的食物供应。季节变化、极端气候、资源矛盾等均增加了食物供给的不确定性,这意味着母体可能长期面临能量摄入不足的风险。如果胎儿完全依赖母体即时的血糖浓度来调控自身的生长,那么母体的短期饥饿就极易导致胎儿发育迟滞,威胁后代存活,降低种群应对环境变化的适应性。
因此,在生死存亡的演化压力下,一个更优的策略被选择出来:让胎儿在程序设计上就“默认”自己处于一个相对资源受限(低糖)的环境,并主动关闭对轻度资源波动的过度应激反应。
TRPV4的乙酰化修饰正是这一策略在分子层面的体现。它相当于为胎儿的生长引擎安装了一个“缓冲器”,使其不会因母体血糖的短期波动而轻易“熄火”,为自己争取每一分生长的可能。
03哲学延伸:生命程序中的“韧性”与“代价”
这一科学发现也引发了超越生物学的哲学性思考。
1、生命的“反脆弱性”: 胎儿并非被动承受低糖环境,而是通过主动的分子修饰,将这一限制条件转化为保障持续生长的稳定背景。这体现了生命系统的一种“反脆弱性”——即在一定的生存压力或环境波动下,反而能激发并固化其核心功能,使其更具韧性,生命的脆弱与坚韧在此辩证统一。
2、演化中的“妥协”与“风险”: 这一保护机制是有代价的。让mTORC1在低糖下持续活跃,可能意味着细胞放弃了部分在营养压力下的精细调控与自我保护能力。这种在发育速度与风险管控之间的权衡,是演化的经典“妥协”。更有趣的是,这种胎儿期的程序设定,是否会在个体出生后留下“印记”?它是否与成年后某些代谢性疾病(如对高热量饮食环境的不适应)的风险存在隐秘联系?这连接起了“健康与疾病发育起源”的DOHaD理论假说。
3、环境与基因的深层对话: TRPV4的乙酰化修饰本身,可能受到表观遗传调控,即环境因素(如母体营养状态)能否影响这一修饰的程度,从而微调胎儿的生长策略?这暗示了环境与基因之间并非简单的指令与执行关系,而是在生命最初期就展开的一场深刻的、多层次的对话。预设的发育程序中,仍留有对所处环境信息交互的接口。
结语
胎儿通过精巧的分子开关(TRPV4乙酰化),屏蔽了低糖对生长发育的抑制信号,捍卫了生长的根本动力。这不仅是保证个体正常发育的生理机制,更是人类作为物种,在残酷的自然选择中演化出的、应对母体营养供给波动的生存智慧。
每一个新生命的孕育,都携带着来自远古的、与饥饿抗争的艰苦记忆与应对策略,这份深植于我们细胞深处的“韧性”,是生命赠予自身的第一份,也是最宝贵的礼物。浙江大学医学院附属第四医院张辰颉医师、复旦大学附属妇产科医院余传金助理研究员为共同第一作者,黄荷凤院士、林圣彩院士、复旦大学附属妇产科医院丁国莲教授、厦门大学生命科学学院张宸崧教授为共同通讯作者。项目得到了国家自然科学基金、国家重点研发计划等多项资助。
文章来源:生命科学开放联盟



